
Pompe Disease
After familiarizing yourself with lysosomal biology, glycogen metabolism, and the latest research on Pompe disease, now you should understand the definition of Pompe Disease.
From the Merriam-Webster Dictionary: an inherited glycogen storage disease that is characterized by the abnormal accumulation of glycogen especially in skeletal and cardiac muscle tissue and that results from a deficiency in a lysosomal enzyme which breaks down glycogen into glucose
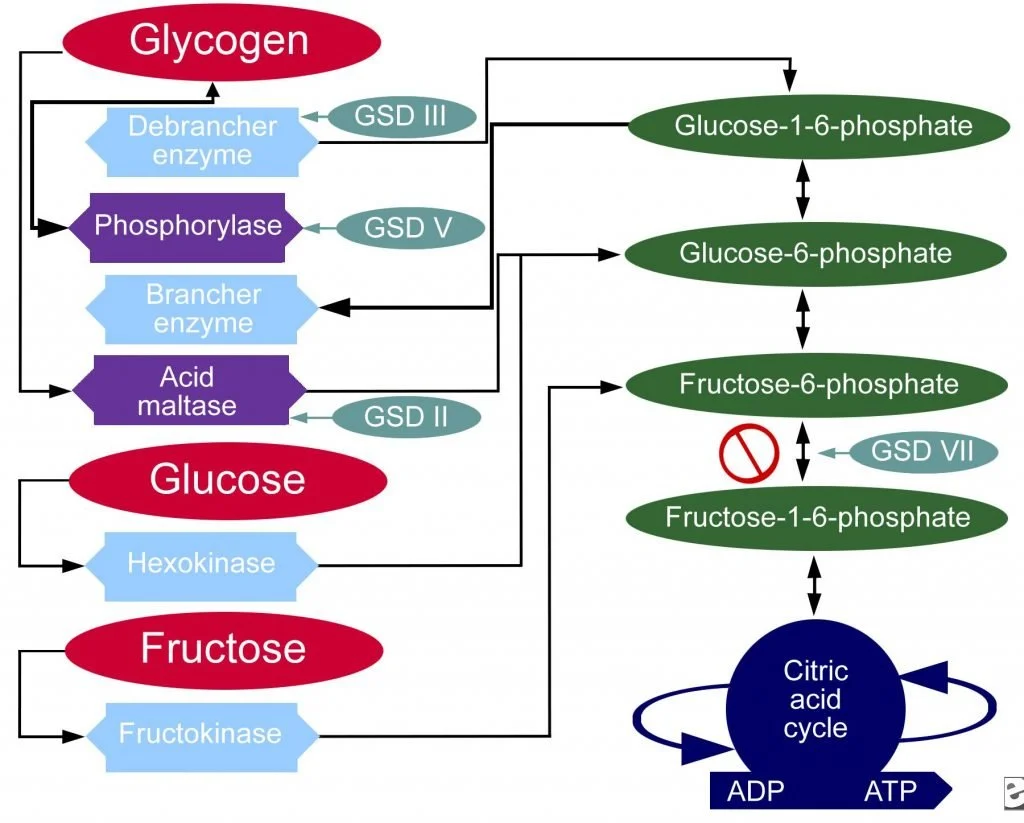
This definition states that “the abnormal accumulation of glycogen” results from a deficiency in a lysosomal enzyme”. All other processes involving glycogen metabolism in the human body remain unchanged. Here is a graphic to show what that means.
Remember that another name for Pompe Disease is Glycogen Storage Disease Type 2, which is abbreviated in the above graphic as GSD II. GSD II does not affect glucose metabolism, fructose metabolism, or the other pathways involved with glycogen metabolism. Since all other glycogen/glucose/fructose metabolism pathways work in Pompe Disease, PWF started asking our medical team a few questions.
Is there a way to not use the glycogen path (see above diagram) that involves acid maltase? Pompe Disease= GSD II = Acid Maltase Deficiency
Is there a faster way to clear out lysosomes in the body once they have gotten stuck on a glycogen molecule?